基于tensorflow框架的基因本体论GO蛋白质embedding的DNN模型
基于tensorflow框架的基因本体论GO蛋白质embedding的DNN模型
- 前言:关于基因本体论GO相关研究论文
- (一)基因本体论GO
- 1.1 模型目的
- 1.2 蛋白质序列
- 1.3 基因本体论(Gene Ontology):
- 1.4 GO术语的构成
- 1.5 文件描述
- 1.6 关于数据集的标签
- (二)用于训练和测试数据的蛋白质embedding
- 2.1 库导入
- (三) 加载数据集
- (四) 加载蛋白质embedding
- (五) 准备数据集
- (六)训练模型
- (七)绘制每个时期的模型损失和精度
- (八)提交模型
- (九) 预测
前言:关于基因本体论GO相关研究论文
1. Gene ontology: tool for the unification of biology
作者:Michael Ashburne
2. The Gene Ontology Resource: 20 years and still GOing strong
作者:The Gene Ontology Consortium
3. The Gene Ontology (GO) database and informatics resource
作者:Michael A. Harris
(一)基因本体论GO
1.1 模型目的
是根据一组蛋白质的氨基酸序列和其他数据预测其功能(又名GO项ID)
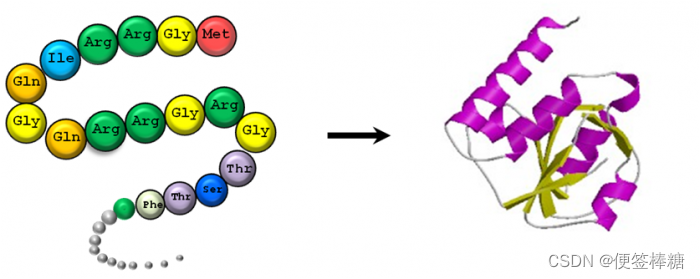
1.2 蛋白质序列
每种蛋白质由数十或数百个按顺序连接的氨基酸组成。序列中的每个氨基酸可以用一个字母或三个字母的代码表示。因此,蛋白质的序列通常被标注为一串字母。
1.3 基因本体论(Gene Ontology):
使用基因本体(GO)来定义蛋白质的功能特性1。基因本体(GO)描述了我们对生物领域的三个方面的理解:
1.分子功能(Molecular Function,MF )
单个的基因产物(包括蛋白质和RNA)或多个基因产物的复合物在分子水平上的活动,比如“催化”,“转运”
需要注意,这里的描述只表示活动,而不指定执行功能的实体(分子或复合物),动作发生的地点,时间或背景
广义上的例子是催化活性和转运蛋白活性。具体的例子是腺苷酸环化酶活性或Toll样受体结合
为避免基因产物名称与其分子功能之间的混淆,GO分子功能通常附加“活性(activity)”一词。比如,蛋白激酶(protein kinase)具有GO分子功能:蛋白激酶活性( protein kinase activity)
2.细胞组分(Cellular Component ,CC)
基因产物在执行功能时所处的细胞结构位置,比如在线粒体,核糖体
需要注意:细胞组分是细胞解刨结构,不指代过程
3.生物过程(Biological Process ,BP)
通过多种分子活动完成的生物学过程,广义上的例子是DNA修复或信号转导。更加具体的例子是嘧啶核苷生物合成过程或葡萄糖跨膜转运(生物学过程不等同于通路。目前,GO没有表示完整的通路信息所需的动力学或依赖性的描述信息)
栗子:
基因产物:细胞色素C(cytochrome c)
分子功能:氧化还原酶活性
细胞组分:线粒体基质
生物过程:氧化磷酸化
1.4 GO术语的构成
1.基本要素
- 唯一标识符(GO ID)和名称:比如GO:0005739,GO:1904659,GO:0016597和线粒体,葡萄糖跨膜转运,氨基酸结合
- 方面: 该术语属于细胞成分,生物过程或分子功能的哪一个。
- 定义:术语的文字描述,以及信息来源的引用。
- 关系:该术语与本体中其他术语的关系。 例如,葡萄糖跨膜转运(GO:1904659)是单糖转运(GO:0015749)。2
官网文档:GO概述
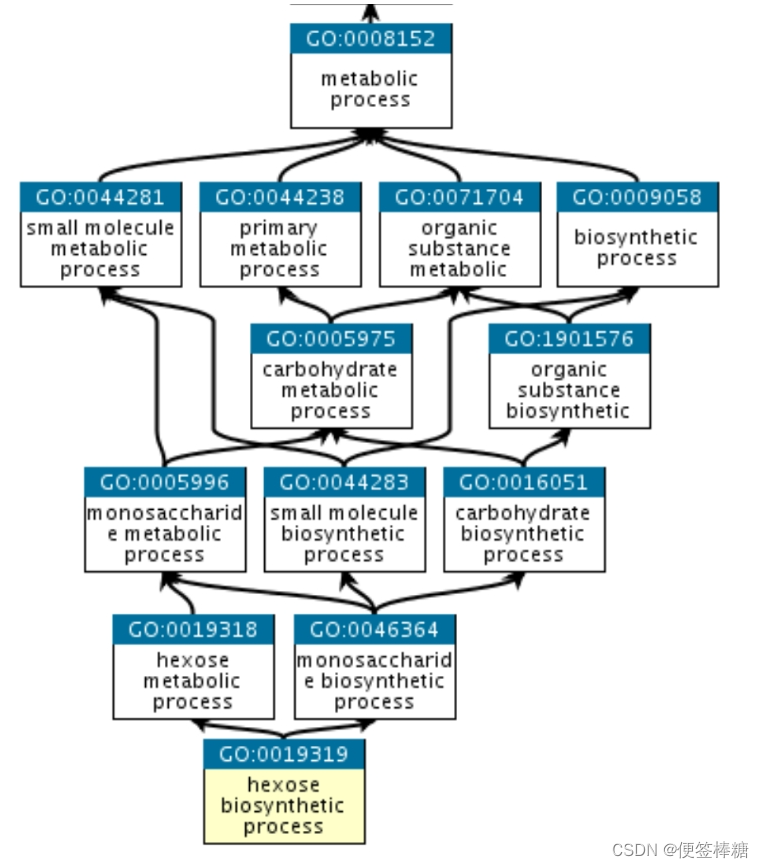
2. GO 图
GO 的结构可以用图来描述,其中每个 GO 项都是一个节点,项之间的关系是节点之间的边。GO 是松散的分层,“子”术语比它们的“父”术语更专业,但与严格的层次结构不同,一个术语可能有多个父术语(请注意,父/子模型不适用于所有类型的关系,请参阅关系文档).例如,生物过程术语己糖生物合成工艺有两个父母,己糖代谢过程和单糖生物合成工艺.这反映了这样一个事实,即生物合成过程是代谢过程的一个亚型,而己糖是单糖的一个亚型。
1.5 文件描述
文件“train_terms.tsv”包含“train_sequences.fasta”中蛋白质的注释术语(基本事实)列表。
在“train_terms.tsv”中:
- 第一列表示蛋白质的UniProt加入ID(唯一蛋白质ID)
- 第二列是“GO术语ID”
- 第三列表示该术语出现在哪个本体中。
1.6 关于数据集的标签
模型的目标是预测蛋白质序列的项(功能)。一个蛋白质序列可以具有许多功能,因此可以分为任意数量的术语。每个术语都由“GO 术语 ID”唯一标识。因此,模型必须预测蛋白质序列的所有“GO Term ID”。这意味着该任务是一个多标签分类问题。

(二)用于训练和测试数据的蛋白质embedding
为了训练机器学习模型,不能直接使用 train_sequences.fasta 中的字母序列。它们必须转换为矢量格式。3
使用蛋白质序列的嵌入来训练模型。(可以认为蛋白质嵌入类似于用于训练NLP模型的词嵌入。)
为了使计算和数据准备更容易,使用预先计算的蛋白质嵌入4
蛋白质embedding是一种机器友好的方法,主要通过其序列捕获蛋白质的结构和功能特征。5
一种方法是训练自定义 ML 模型6。由于该数据集使用氨基酸序列(这是一种标准方法)表示蛋白质,因此可以使用任何公开可用的预训练蛋白质嵌入模型来生成嵌入。7
2.1 库导入
import tensorflow as tf
import pandas as pd
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt# Required for progressbar widget
import progressbar
print("TensorFlow v" + tf.__version__)
print("Numpy v" + np.__version__)

(三) 加载数据集
加载文件“train_terms.tsv”,其中包含蛋白质的注释术语(函数)列表。将提取标签,又名“GO 术语 ID”,并为蛋白质嵌入创建一个标签数据帧8。
train_terms = pd.read_csv("input/cafa-5-protein-function-prediction/Train/train_terms.tsv",sep="\t")
print(train_terms.shape)
train_terms 数据帧由 3 列和 5363863 个条目组成。我们可以使用以下代码打印出前 3 个条目来查看数据集的所有 5 个维度
train_terms.head()
( )]
)]
(四) 加载蛋白质embedding
使用Rost Lab的T5蛋白质语言模型,用于训练的蛋白质嵌入记录在“train_embeds.npy”中,相应的蛋白质ID在“train_ids.npy”中可用。
将 ‘train_ids.npy’ 中包含的训练数据集中蛋白质嵌入的蛋白质 id 加载到 numpy 数组中。
train_protein_ids = np.load('/input/t5embeds/train_ids.npy')
print(train_protein_ids.shape)
train_protein_ids[:5]
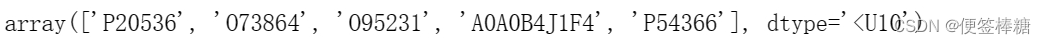
将文件加载为 numpy 数组后,将它们转换为 Pandas 数据帧。
每个蛋白质embedding都是长度为1024的载体。创建结果数据帧,以便有 1024 列来表示向量中 1024 个位置中每个位置的值。
train_embeddings = np.load('/kaggle/input/t5embeds/train_embeds.npy')# Now lets convert embeddings numpy array(train_embeddings) into pandas dataframe.
column_num = train_embeddings.shape[1]
train_df = pd.DataFrame(train_embeddings, columns = ["Column_" + str(i) for i in range(1, column_num+1)])
print(train_df.shape)
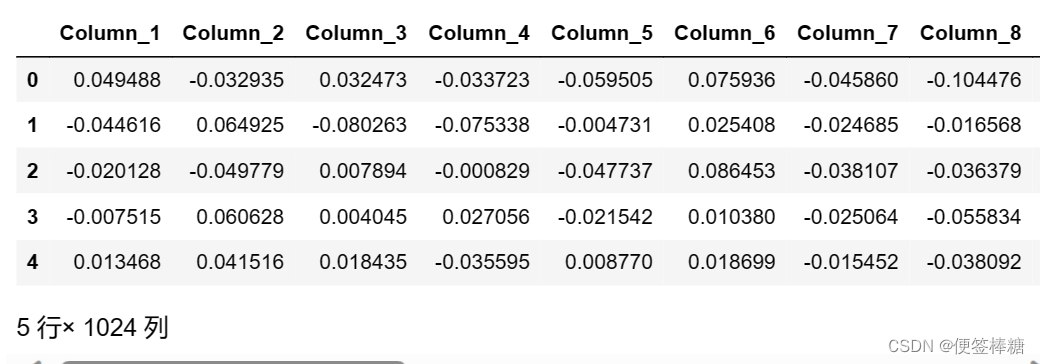
(五) 准备数据集
从“train_terms.tsv”文件中提取所有需要的标签(“GO 术语 ID”)。总共有超过 40,000 个标签。
为了简化模型,将选择最常见的 1500 个 ‘GO 术语 ID’ 作为标签。
在“train_terms.tsv”中绘制最常见的 100 个“GO 术语 ID”。
# Select first 1500 values for plotting
plot_df = train_terms['term'].value_counts().iloc[:100]figure, axis = plt.subplots(1, 1, figsize=(12, 6))bp = sns.barplot(ax=axis, x=np.array(plot_df.index), y=plot_df.values)
bp.set_xticklabels(bp.get_xticklabels(), rotation=90, size = 6)
axis.set_title('Top 100 frequent GO term IDs')
bp.set_xlabel("GO term IDs", fontsize = 12)
bp.set_ylabel("Count", fontsize = 12)
plt.show()
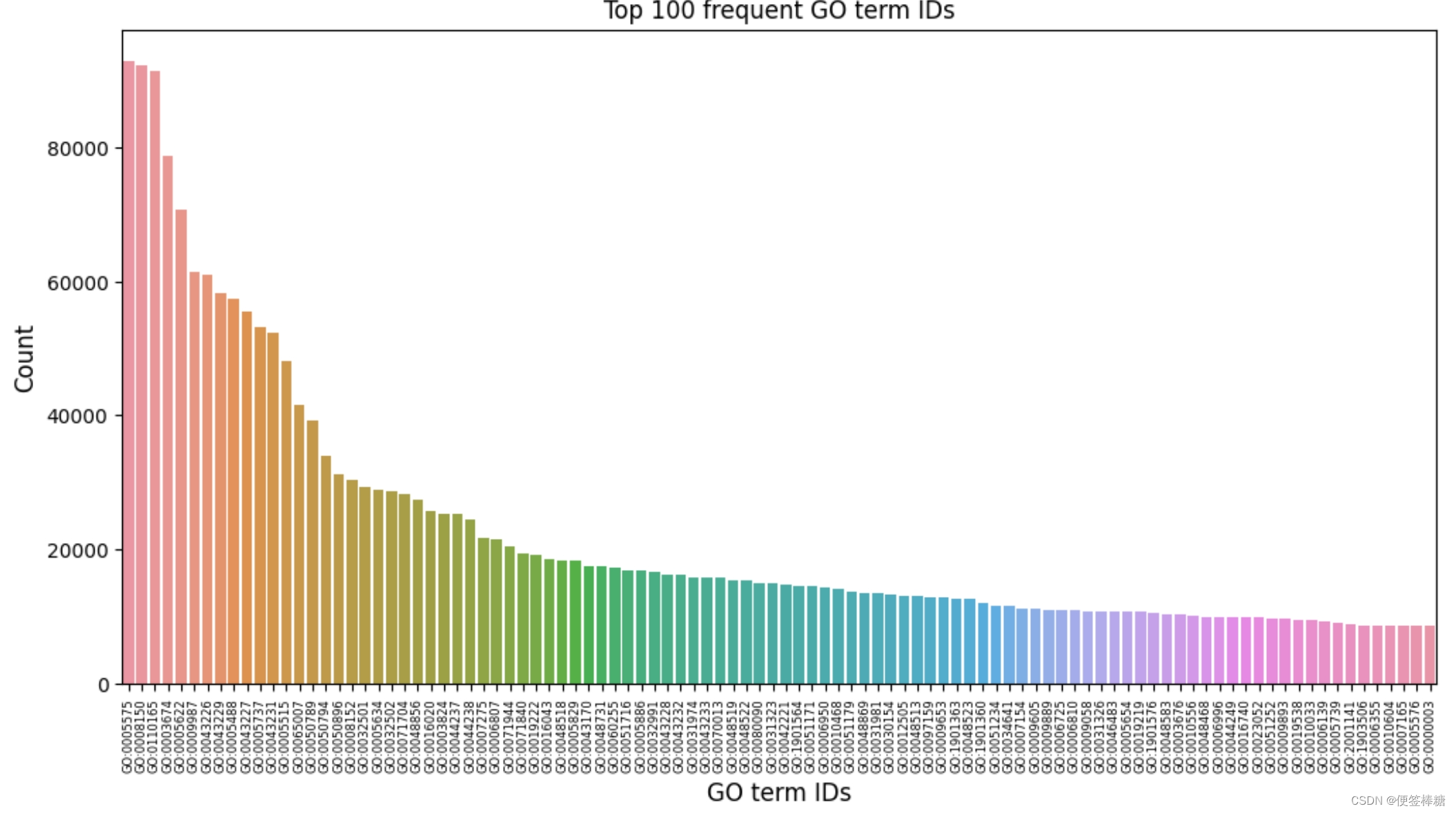
将前 1500 个最常用的 GO 术语 ID 保存到一个列表中。
# Set the limit for label
num_of_labels = 1500# Take value counts in descending order and fetch first 1500 `GO term ID` as labels
labels = train_terms['term'].value_counts().index[:num_of_labels].tolist()
使用选定的“GO 术语 ID”过滤训练术语来创建一个新的数据帧
# Fetch the train_terms data for the relevant labels only
train_terms_updated = train_terms.loc[train_terms['term'].isin(labels)]
使用饼图绘制新train_terms_updated数据框中的纵横比值:
pie_df = train_terms_updated['aspect'].value_counts()
palette_color = sns.color_palette('bright')
plt.pie(pie_df.values, labels=np.array(pie_df.index), colors=palette_color, autopct='%.0f%%')
plt.show()
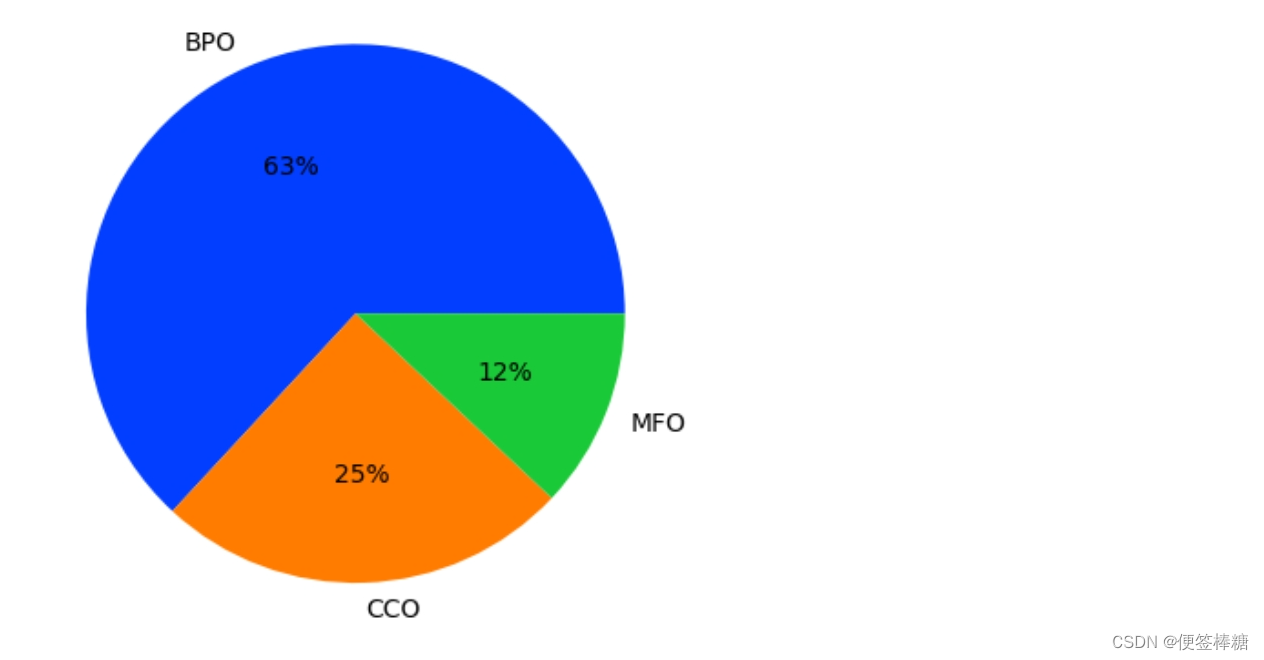
63% “GO术语Id”都以BPO(生物过程本体)为主要因素。
由于这是一个多标签分类问题,因此在标签数组中,使用 1 或 0 表示蛋白质 ID 的每个 Go Term ID 的存在与否。
首先,为标签创建一个所需大小的 numpy 数组 ‘train_labels’。要使用适当的值更新 ‘train_labels’ 数组,遍历标签列表。
# Setup progressbar settings.
# This is strictly for aesthetic.
bar = progressbar.ProgressBar(maxval=num_of_labels, \widgets=[progressbar.Bar('=', '[', ']'), ' ', progressbar.Percentage()])# Create an empty dataframe of required size for storing the labels,
# i.e, train_size x num_of_labels (142246 x 1500)
train_size = train_protein_ids.shape[0] # len(X)
train_labels = np.zeros((train_size ,num_of_labels))# Convert from numpy to pandas series for better handling
series_train_protein_ids = pd.Series(train_protein_ids)# Loop through each label
for i in range(num_of_labels):# For each label, fetch the corresponding train_terms datan_train_terms = train_terms_updated[train_terms_updated['term'] == labels[i]]# Fetch all the unique EntryId aka proteins related to the current label(GO term ID)label_related_proteins = n_train_terms['EntryID'].unique()# In the series_train_protein_ids pandas series, if a protein is related# to the current label, then mark it as 1, else 0.# Replace the ith column of train_Y with with that pandas series.train_labels[:,i] = series_train_protein_ids.isin(label_related_proteins).astype(float)# Progress bar percentage increasebar.update(i+1)# Notify the end of progress bar
bar.finish()# Convert train_Y numpy into pandas dataframe
labels_df = pd.DataFrame(data = train_labels, columns = labels)
print(labels_df.shape)
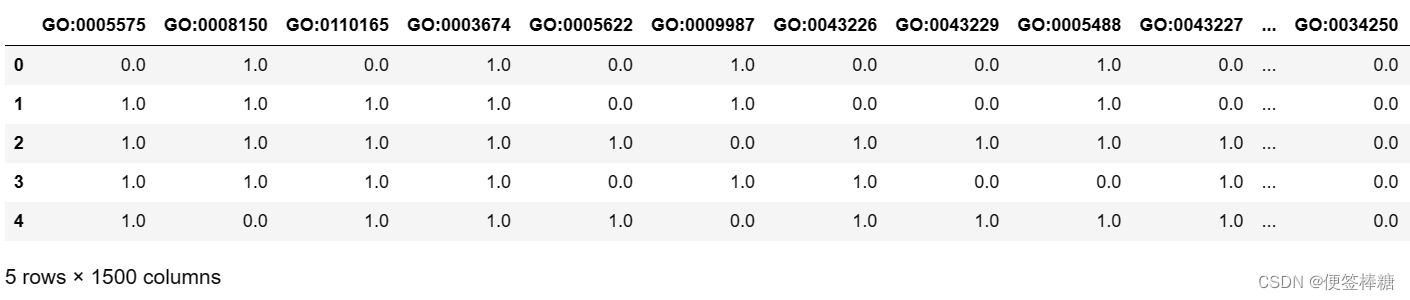
最终标签数据帧 (‘label_df’) 由 1500 列和 142246 个条目组成。打印出前 5 个条目,从而查看数据集的所有 1500 个维度
labels_df.head()
(六)训练模型
使用 Tensorflow 来训练带有蛋白质嵌入的深度神经网络
INPUT_SHAPE = [train_df.shape[1]]
BATCH_SIZE = 5120model = tf.keras.Sequential([tf.keras.layers.BatchNormalization(input_shape=INPUT_SHAPE), tf.keras.layers.Dense(units=512, activation='relu'),tf.keras.layers.Dense(units=512, activation='relu'),tf.keras.layers.Dense(units=512, activation='relu'),tf.keras.layers.Dense(units=num_of_labels,activation='sigmoid')
])# Compile model
model.compile(optimizer=tf.keras.optimizers.Adam(learning_rate=0.001),loss='binary_crossentropy',metrics=['binary_accuracy', tf.keras.metrics.AUC()],
)history = model.fit(train_df, labels_df,batch_size=BATCH_SIZE,epochs=5
)

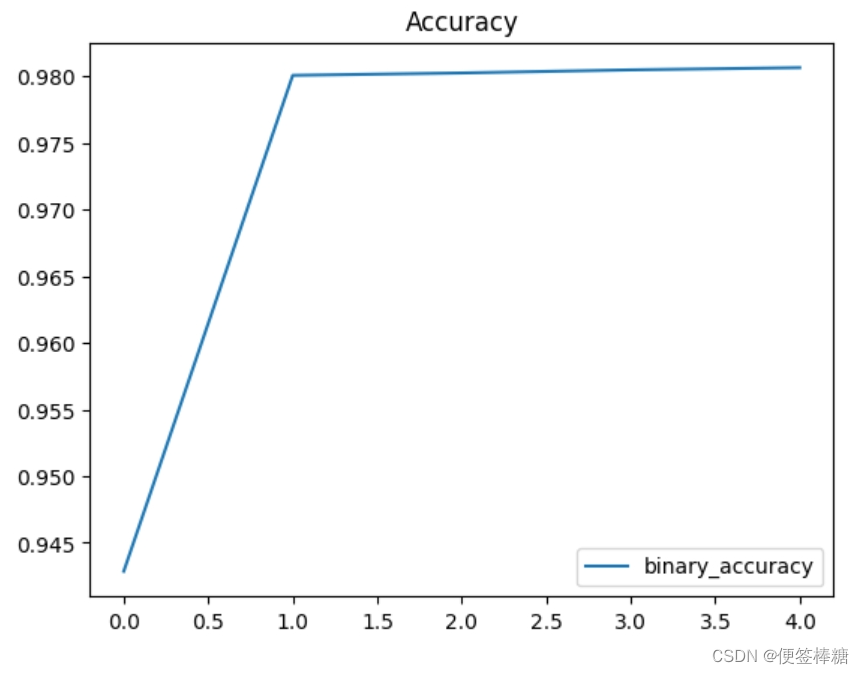
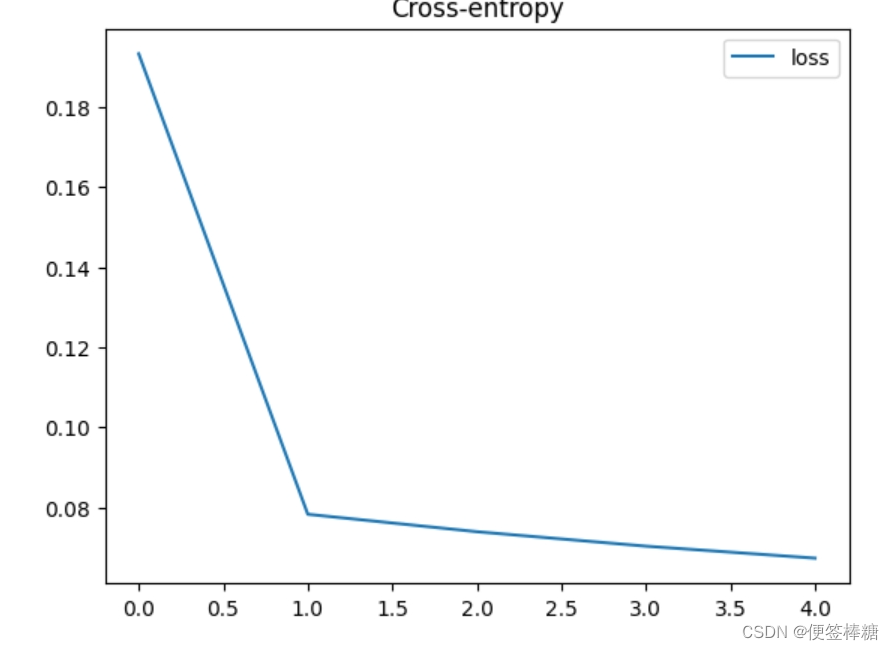
(七)绘制每个时期的模型损失和精度
history_df = pd.DataFrame(history.history)
history_df.loc[:, ['loss']].plot(title="Cross-entropy")
history_df.loc[:, ['binary_accuracy']].plot(title="Accuracy")
(八)提交模型
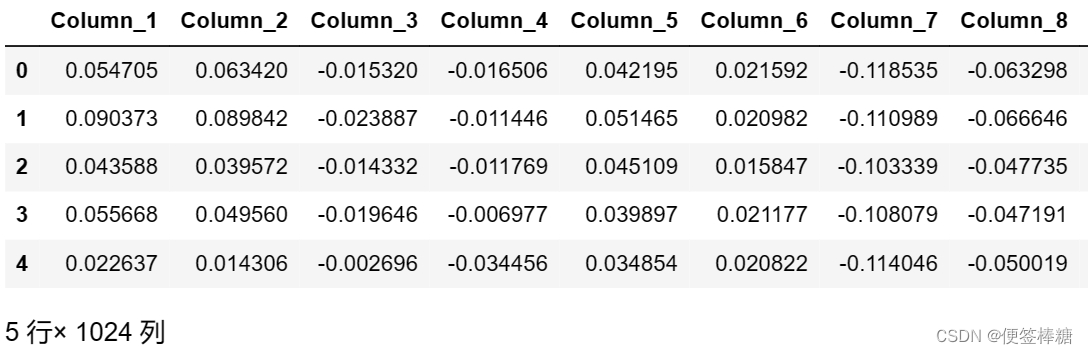
test_embeddings = np.load('/kaggle/input/t5embeds/test_embeds.npy')# Convert test_embeddings to dataframe
column_num = test_embeddings.shape[1]
test_df = pd.DataFrame(test_embeddings, columns = ["Column_" + str(i) for i in range(1, column_num+1)])
print(test_df.shape)

test_df.head()
(九) 预测
predictions = model.predict(test_df)
# Reference: https://www.kaggle.com/code/alexandervc/baseline-multilabel-to-multitarget-binarydf_submission = pd.DataFrame(columns = ['Protein Id', 'GO Term Id','Prediction'])
test_protein_ids = np.load('/kaggle/input/t5embeds/test_ids.npy')
l = []
for k in list(test_protein_ids):l += [ k] * predictions.shape[1] df_submission['Protein Id'] = l
df_submission['GO Term Id'] = labels * predictions.shape[0]
df_submission['Prediction'] = predictions.ravel()
df_submission.to_csv("submission.tsv",header=False, index=False, sep="\t")
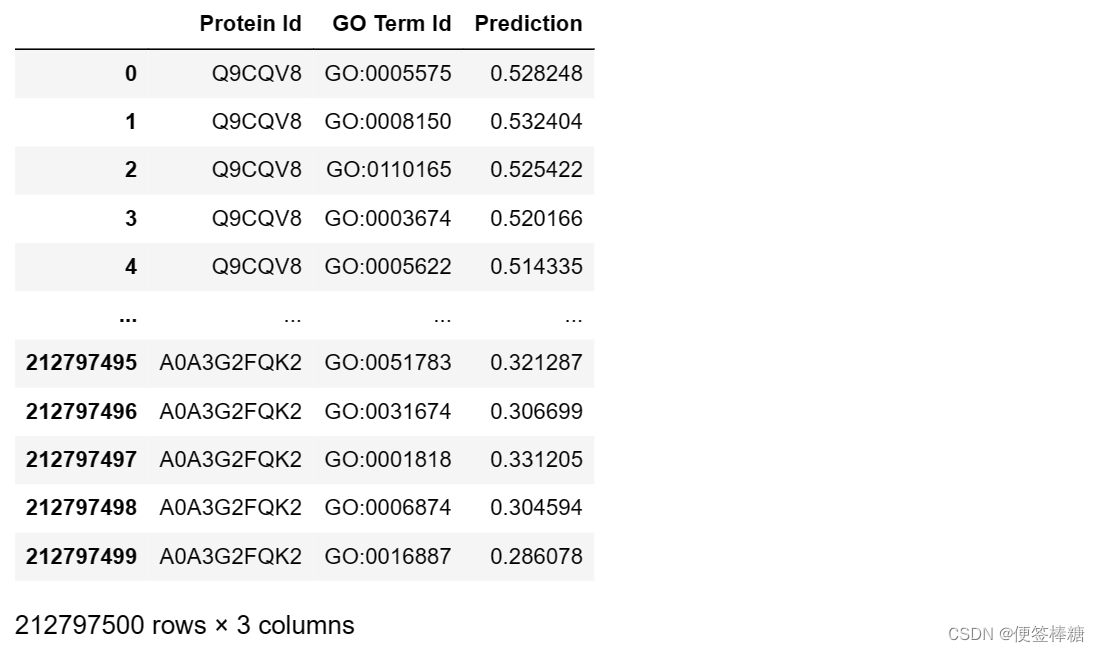
df_submission
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000 May;25(1):25-9. doi: 10.1038/75556. PMID: 10802651; PMCID: PMC3037419. ↩︎
https://zhuanlan.zhihu.com/p/99789859 ↩︎
Heinzinger, M., Bagheri, M., & Kelley, L. A. (2017). Deep learning for predicting protein backbone structure from amino acid sequences. Scientific reports, 7(1), 1-11. ↩︎
https://zhuanlan.zhihu.com/p/593730989 ↩︎
https://blog.csdn.net/weixin_43841338/article/details/103171724 ↩︎
AlQuraishi, M. (2019). End-to-end differentiable learning of protein structure. Cell Systems, 8(4), 292-301. ↩︎
https://zhuanlan.zhihu.com/p/498353259 ↩︎
Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., … & Zisserman, A. (2020). Improved protein structure prediction using potentials from deep learning. Nature, 577(7792), 706-710. ↩︎
本文来自互联网用户投稿,文章观点仅代表作者本人,不代表本站立场,不承担相关法律责任。如若转载,请注明出处。 如若内容造成侵权/违法违规/事实不符,请点击【内容举报】进行投诉反馈!